Introduction générale I. Concepts et définitions L'objet de cet ouvrage est l'analyse des flux métaboliques de substances endogènes telles que des hormones, des médiateurs chimiques, des protéines, des lipides, etc. produits par l'organisme lui-même. Les méthodes utilisées sont dérivées de celles de la pharmacocinétique qui traite du devenir des substances exogènes étrangères à l'organisme que sont les médicaments, d'où la place importante accordée à la pharmacocinétique tout au long des pages suivantes. D'un point de vue cinétique médicaments et marqueurs de substances endogènes sont identiques. Le "médicament" (medicine, drug chez les anglo-saxons) est une substance thérapeutique dont le but est d’améliorer les conditions physiologiques d’un organisme ayant subi une altération de ces conditions, par rapport à un organisme sain ou supposé comme tel. Le mot "drogue" en français (drugs en anglais) exprime l’aspect négatif d’une substance. Les manipulations et l’étude des médicaments sont définis par des termes organisés autour du mot grec "j a r m a k o n " (pharmacon=médicament). On distingue la "Pharmacie" qui est l’échoppe du pharmacien où les médicaments sont préparés, stockés et vendues. La "Pharmacopée" (de j a r m a k o n et p o i e i n = faire) désigne l’art de préparer les médicaments. Ce terme désigne aussi l’ensemble des spécialités délivrées par le pharmacien et dont la liste officielle constitue le "Codex medicamentarius" (codex = recueil de lois). Les médicaments sont présentés sous différents aspects ou "formes galéniques" (de Galien médecin latin) |
|||
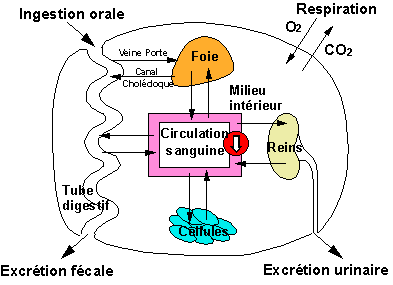
Figure 1 : Schéma des échanges physiologiques chez un mammifère. |
|
||
| La
"Pharmacologie" (de j a r m a k o n et l o g o V = étude) s’intéresse au médicament, et plus
généralement à toute substance capable d’agir sur l’organisme, entre le
moment où il est administré et l’obtention de l’effet. Elle est subdivisée en
trois étapes correspondant à trois phases subies par le médicament au cours du
temps : + la "phase biopharmaceutique" correspond à la mise à disposition du médicament pour l’organisme (désintégration de la forme solide dans l’estomac par exemple, désagrégation en particules fines pour favoriser la dissolution et le transfert de l’agent actif à travers les membranes biologiques). + la "phase pharmacocinétique" correspond au devenir in vivo du médicament (absorption, distribution, élimination) en fonction du temps (Sheppard, 1962; Atkins, 1969; Shipley and Clark, 1972; Chevallier, 1984; Aiache et coll., 1990; Magot et Champarnaud, 1990). Par exemple, un médicament administré par voie orale ou anale est absorbé à partir des sites gastro-intestinaux. Il est transporté par la veine porte hépatique au foie avant d’être dispersé dans la circulation systémique. Au cours de ces transferts, plusieurs facteurs peuvent affecter la quantité de substance active qui entre dans la circulation et la vitesse à laquelle elle l’atteint. La "biodisponibilité" représente la fraction de la substance active qui atteint la circulation sanguine générale. Dans la circulation sanguine, la vitesse de transfert du médicament aux sites d’action (récepteurs membranaires des cellules visées) dépend de sa liaison plus ou moins forte aux protéines plasmatiques et aux récepteurs extravasculaires. Ces interactions avec les protéines circulantes et les récepteurs cellulaires affectent la distribution du médicament entre les tissus et le sang, son niveau de toxicité ou d’innocuité, sa durée d’action, etc... Le terme final pharmacocinétique est l’élimination du médicament, intact ou sous forme de métabolites, par le rein et/ou le foie avec possibilité de "cycle entérohépatique". L’élimination du principe actif est caractérisée par sa constante de vitesse d’élimination et sa demi-vie. + la "phase pharmacodynamique" est le résultat de l’interaction récepteur-principe actif du médicament administré. De nombreux facteurs peuvent modifier la sensibilité du récepteur au médicament, tels que l’âge, le sexe, l’état pathologique, d’autres médicaments, les rythmes biologiques, etc... Il est clair que le déroulement des processus dans chaque phase conditionne la phase suivante. Une dissolution très lente du médicament par exemple peut avoir pour conséquence une répartition prolongée dans le temps et un temps d’action plus long ou au contraire un contact prolongé avec des enzymes destructeurs. Que le médicament soit administré par voie intraveineuse (IV), intramusculaire (IM), orale ou anale, les étapes pharmacocinétiques et pharmacodynamiques sont de même nature, mais les constantes de temps et de concentration sont modifiées par le mode d’administration. La "Pharmacocinétique" qui est un élément de ce cours n’est donc pas indépendante. Elle est conditionnée par la "Biopharmaceutique" et conditionne à son tour la "Pharmacodynamique". Un développement récent de la pharmacocinétique est la "Chronopharmacocinétique" associée au concept de "Chronopharmacologie". Dans sa thèse de médecine, soutenue en 1814, Virey (cité par Reinberg et al., 1991) pensait que l’effet des médicaments variait en fonction de l’heure de leur administration mais ce n’est qu’à partir des années 1960 que cette hypothèse a été expérimentalement vérifiée et que des applications pratiques ont été proposées (Bruguerolle, 1987; Reinberg et al., 1991). Par exemple, chez l’Homme, l’acide acétylsalicylique (aspirine) offre une meilleure biodisponibilité (biopharmaceutique et absorption gastro-intestinale) lorsqu’il est ingéré le matin qu’à un autre moment de la journée. Chez la femme l’absorption du salicylate de sodium est maximale vers le milieu du cycle menstruel. Chez la rate, la théophylline est 5 fois mieux absorbée en fin de cycle oestrien qu’en début de cycle (Bruguerolle, 1987). De nombreux facteurs sont susceptibles de modifier le profil cinétique d'une substance active (alternance entre repas et jeûne, modifications du pH urinaire, variations du débit sanguin hépatique, etc...) Les principes de la Pharmacocinétique ne sont pas limités à l’étude cinétique des médicaments. Depuis plusieurs décennies, les physiologistes les ont appliqués à l’étude des substances endogènes grâce à des molécules radioactives utilisées comme marqueurs identiques aux molécules non radioactives endogènes (Hevesey, 1923; Hevesey and Hofer, 1934; Schoenheimer and Rittenberg, 1935 et 1939; Zilversmit et al., 1943; Brownell, 1951; Riggs, 1952; Sheppard, 1962; Tait and Burnstein, 1964; Atkins, 1969; Chevallier, 1972 et 1984; Shipley and Clark, 1972; Magot et Champarnaud, 1990). Nous traiterons donc de la pharmacocinétique élargie à toutes les molécules (exogènes et surtout endogènes) que l'on a désignée par "Analyse des flux métaboliques" et de l’usage des molécules radioactives comme marqueurs ou traceurs. |
|||

|
|||